Our lab uses a multidisciplinary approach to address fundamental aspects of protein folding, misfolding and aggregation. In addition to define the basic mechanistic principles underlying these processes, we aim to understand how their deregulation leads to the onset of human conformational diseases and to develop innovative therapeutics to target these pathologies. Moreover, this knowledge should allow us to design and produce novel and better protein-based biopharmaceuticals as well as the development of new self-assembled materials for nanotechnology applications.
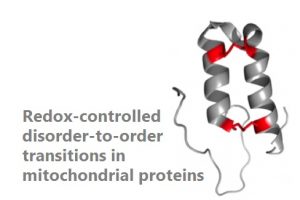

For most proteins, biological function requires folding into a unique structure, in which they remain during their lifetime. There is, however, an emerging class of proteins that adopt a specific structure to function, but are otherwise disordered, the so-called conditionally disordered proteins (CDPs). The structure and activity of CDPs is tightly regulated by the environment. Some CDPs, use oxidation of their redox-sensitive cysteines to reversibly convert large disordered regions into defined structural domains. This is the case of proteins synthesized on cytosolic ribosomes and targeted to the intermembrane space (IMS) of mitochondria as reduced and disordered species. They become oxidized and folded into their functional forms only after entering this organelle. This redox-controlled folding, acts as a folding-trap that drives their translocation to the IMS. We use an integrated structural approach to decipher the molecular determinants of these massive structural rearrangements, which determine the function of mitochondrial proteins linked to diseases like Parkinson’s, Huntington’s or cardiomyopathies
Parkinson's disease (PD) is the second most common neurodegenerative disorder and is still incurable. PD is associated with the death of dopaminergic neurons in the brain. There is substantial evidence supporting the aggregation of the protein a-Synuclein (a-Syn) as a key event in pathogenesis of PD, emerging thus as a privileged therapeutic target. We have recently identified and designed small compounds and peptides able to inhibit a-Syn aggregation with extremely high potency in vitro and in animal models. We aim to develop these molecules into lead compounds for the therapeutics of PD.
Early detection of PD is a long pursued objective. A predictive test would revolutionize clinical care, research and treatment. Biochemical analysis evidenced an elevation of a-Syn aggregates in the biofluids of patients, suggesting that this feature might be used as a diagnostic biomarker for PD. We aim to develop an orthogonal approach towards the development of a sensitive automated diagnostic assay based on the specific detection of early a-Syn aggregates in biofluids.
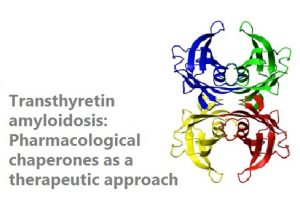
Transthyretin amyloidosis (ATTR) is the most common form of familial amyloidosis. In ATTR, destabilizing mutations in the protein transthyretin (TTR) causes amyloid fibres to build up, which, depending on the mutation, are deposited in different organs, such as the brain, the nerves or the myocardium, causing them to malfunction and bringing the various forms of the disease. To prevent disease progress, a liver transplant or heart transplant is needed. However, the use of pharmacologic chaperones that stabilize the structure of TTR is emerging as a non-invasive therapeutic means to halt the disease. In this context, we have repurposed a molecule originally intended to treat Parkinson’s, as perhaps, the most effective drug for these diseases, already in clinical trials. We pursue both to test its efficacy for previously unaddressed forms of ATTR, with special emphasis of those occurring in the brain, and to use structure based design to generate second generation, more powerful, drugs
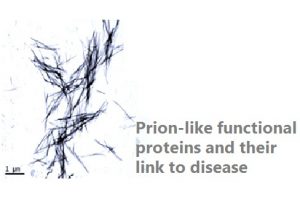
Amyloids become infectious in prion diseases. Nevertheless, not all prion proteins are disease-related; in yeast, they help for environmental adaptation. Most yeast prions contain a low complexity (LC) prion domain responsible for their self-assembly and propagation. Proteins from other organisms such as bacteria, plants and humans do also bear prion-like domains indicating that their aggregated state might also be beneficial for the cell. Human prion-like proteins are involved in the formation of membraneless intracellular compartments through liquid-liquid phase separation using their LC domains to favor functional interactions between specific partners. However, these proteins are inherently aggregation-prone and the liquid state can revert into an aberrant solid state responsible for several pathologies including inflammatory and neurodegenerative disorders and cancer. We have recently developed a set of novel algorithms aimed to uncover novel proteins with prion-like behavior in order to characterize the functional pathways in which they are involved as well as their association to disease.
The extraordinary stability and tunable assembly of amyloid fibrils make them very attractive targets in nanotechnology. Most efforts so far have been focused on the use of short synthetic peptides as the bioactive components of such materials, and an analogous approach for inducing globular proteins to assemble into functional nanofibres has been much less explored. The main limitations to create mono- or multi-component nanofibres that contain functional globular proteins come from the requirement to prevent their aggregation during expression, to maintain them in a soluble state during purification and storage, and to be able to induce their assembly at a desired time and place. We aim to exploit our combined computational/experimental expertise to design and produce new molecules fulfilling these properties for a range of biomedical and biotechnological applications, including enzyme catalysis, biosensors, electronics, tissue engineering, drug delivery and immunotherapy.
The fast development of protein therapeutics- monoclonal antibodies, replacement enzymes and hormones- is providing improved therapies for a wide range of human diseases, taking advantage of their high specificity towards their targets. One of the major challenges that one should face during the development of protein-based biopharmaceuticals is their inherent propensity to aggregate. Indeed, protein therapeutic agents are both stored and typically administered at very high concentrations. Under these conditions they can easily aggregate, impacting the product's developability, stability, formulation, and immunogenicity. Traditionally, attempts to improve protein solubility have exploited experimental trial and error approaches. However, they are expensive, difficult to perform and time consuming. We have overcome these limitations developing AGGRESCAN3D, a very efficient computational tool for the automated design of protein structures displaying improved solubility, without compromising their thermodynamic stability and function. We aim to use this approach to assist the production of novel and better protein therapeutics.
