 https://www.frontiersin.org/articles/10.3389/fnmol.2020.582488/full
https://www.frontiersin.org/articles/10.3389/fnmol.2020.582488/full
Introduction
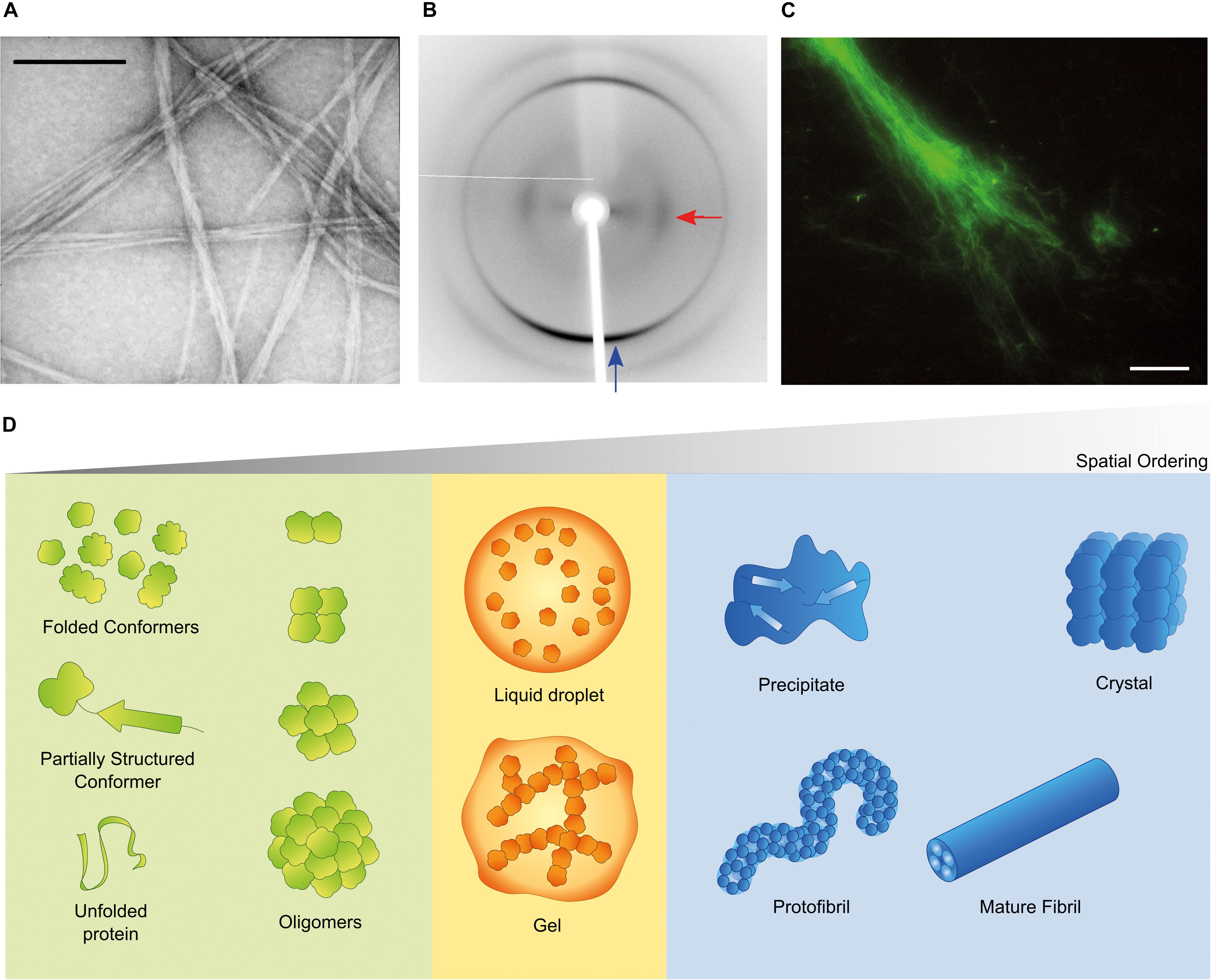
Aggregation of misfolded proteins and peptides is associated with common and rare neurodegenerative disorders and with amyloidoses (Aguzzi and O’Connor, 2010; Chiti and Dobson, 2017; Benson et al., 2018). Classical neuropathological hallmarks such as neurofibrillary tangles in Alzheimer’s disease (AD), Lewy bodies in Parkinson’s disease (PD), or inclusion bodies in Huntington’s disease, have as main constituent characteristic polypeptides aggregated in the form of insoluble highly-ordered structures named amyloid fibrils (Westermark et al., 2007; Gremer et al., 2017). The definition of amyloid encompasses morphological (Figure 1A), structural (Figure 1B) and histological (Figure 1C) aspects. Amyloid polymorphs are associated with different clinical sub-types of the same disease (Guo et al., 2013; Qiang et al., 2017; Goedert et al., 2018), and they exhibit structural differences that can be characterized at atomic resolution using solid-state nuclear magnetic resonance with magic angle spinning (ss-NMR) and cryo-electron microscopy (cryo-EM) techniques (Fitzpatrick et al., 2013; Qiang et al., 2017; Rasmussen et al., 2017; Iadanza et al., 2018b). When the amyloid fold is exploited by nature for functional purposes, the structural variability is less evident than in disease-related aggregates, thus suggesting that functional amyloids are the result of naturally evolved amino acid sequences (Otzen and Riek, 2019). The analogy of a low-energy “black hole” has been used to illustrate the propensity of proteins to form amyloid fibrils after incomplete native folding or if subjected to appropriate denaturing conditions; under mildly denaturing conditions, native interactions and intrachain interactions are in competition, so that the occurrence of transiently stable, non-native conformations may trigger amyloid fibril formation (Zheng et al., 2013). However, there are other energetically stable structures into which proteins self-assemble irrespectively of, prior to, or concomitantly with the formation of amyloid fibrils (Figure 1D). Of these, the soluble aggregates of amyloidogenic proteins attract a particular interest justified by the cytotoxic properties attributed to amyloid-β (Aβ) oligomers in AD (Ahmed et al., 2010; Yang et al., 2017) and α-synuclein oligomers in PD (Lorenzen et al., 2014; Ingelsson, 2016). Liquid-liquid phase separation is now recognized as having a central role in cell physiology and disease (Shin and Brangwynne, 2017). For example, membraneless compartments of concentrated proteins/nucleic acids are implicated in diverse processes, including RNA metabolism, ribosome assembly, DNA repair and intracellular signaling (Banani et al., 2017). When the capacity of protein quality control by the proteasome is exceeded, misfolded proteins are sequestered into intracellular compartments such as the aggresome, a perinuclear deposit destined to autophagy (Johnston et al., 1998), or the CytoQ and INQ, which are deposition sites of misfolded proteins in the cytosol and the nucleus, respectively (Miller et al., 2015). On the other hand, a neuropathological hallmark of sporadic and inherited forms of amyotrophic lateral sclerosis and frontotemporal dementia is the deposition of poorly soluble assemblies of mutated RNA-binding proteins in the nucleus and cytoplasm of neurons (Patel et al., 2015). The pathogenic mutants are characterized by a diminished ability to reiteratively shift between dispersed and condensed phases consisting of dense liquids and gels (Murakami et al., 2015). The physical properties of the different polypeptide assemblies (summarized in Figure 1D) are determined by the degrees of molecular and supramolecular order. Although denser liquid phases relax more slowly in response to shear deformation, they still lack the long-range translational order characteristic of solids (Falahati and Haji-Akbari, 2019). The maximal organization provided by crystal lattices allows the structure of folded proteins to be solved using X-ray crystallography. Naturally occurring microcrystals are also used by living cells for protein storage, protection and stabilization (Schönherr et al., 2018), whereas in crystallopathies such as eosinophilic inflammation, protein crystals have been reported as promising drug targets (Persson et al., 2019).